Research OverviewCellular viability and function require exquisite spatial organization and temporal coordination over a broad range of length- and time- scales. At the nanoscale, proteins must be allowed to explore appropriate ensembles of conformations in order to guarantee the right interactions with their environment and with each other, and yet, this level of organization would be meaningless were it not also the case that the overall spatial distribution of proteins and other cellular components is tightly regulated at the microscale. Moreover, events at every scale are strongly coupled: a single mutation in a protein sequence can lead to misfolding, aggregation, and inclusion formation that is ultimately visible under a microscope. The coalescence of such an inclusion can, in turn, so dramatically alter the load experienced by cellular protein folding quality control machinery that the conformational states of many proteins may be altered. Until recently, limitations in theoretical, computational, and experimental methods prevented researchers from investigating systems of interest on multiple scales simultaneously: scientists either attempted to crystallize a fragment of a single protein, or observed cellular behavior in ignorance of many events occurring at the molecular level. Simultaneous advances in a variety of technological areas now allow significant new discoveries in the field of cell biology to be achieved. Foremost among these, super-resolution fluorescent microscopy has been adapted to the examination of protein interactions in live cells with molecular-scale precision. For the first time, it is possible to correlate genuinely biological phenomena at the level of the whole cell with specific molecular events that cause them, shifting the fundamental paradigm of molecular biology out of the test tube and into the cell where life takes place.
Our goal is to provide the first complete spatial map of protein folding as it takes place in vivo, focusing on cell culture and yeast models of neurodegenerative disease as systems for studying protein folding quality control. The many different pathways -- such as synthesis, ubiquitination, or interaction with chaperones -- that regulate and modulate folding have been extensively characterized in vitro, and yet in order to comprehend the coordinated action of these different components of the cellular machinery, it is essential to observe how they combine to form a dynamic and highly organized quality control super-structure that extends throughout the cell. The dynamic compartmentalization of a whole host of biochemical processes (ranging from protein translation and degradation to RNA quality control and viral assembly) remained invisible to conventional biochemical methods, which do not inspect the cell optically before lysis and homogenization. Now, for the first time, imaging techniques such as super-resolution imaging and laser manipulation have emerged as indispensable tools for resolving various distinct populations of macromolecules and correlating each molecule's trajectory through space with biochemical interactions and changes in conformational state. The application of new high-resolution, dynamic imaging technologies enable us to relate biochemical and genetic information to a spatio-temporal map of events in the process of cellular protein folding quality control. |
 The crowded cytosolic environment  All proteins have to fold 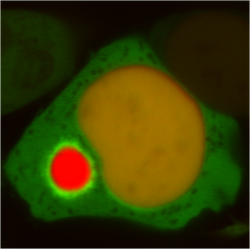 A cell with a polyglutamine Huntingtin inclusion 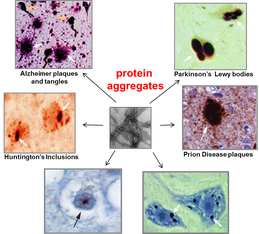 Aggregation causes neurodegenerative diseases |
Projects
Replicative Rejuvenation and Aging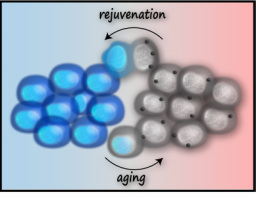
Two important discoveries point to essential clues in the search for the mechanistic basis of aging: replicative rejuvenation and induced pluripotency, or reprogramming to iPSC. Studies of replicative aging have shown that a robust mechanism for aging avoidance promotes the “replicative rejuvenation” of individual cells, from prokaryotes, to budding yeast, mammalian cell lines, and even differentiating stem cells. These cells mitigate the causes and consequences of aging by asymmetrically partitioning the aging determinants during mitosis. Although several of these factors have been identified (oxidatively damaged proteins, old mitochondria, circular DNA, and misfolded proteins, among others), most factors, as well as the mechanism governing asymmetric inheritance, remain a mystery. Understanding the mechanism of replicative rejuvenation will offer definitive insight into the determinants of aging and the interplay between these determinants and disease accumulation.
Why do aggregates affect neurons?
One of the central questions in neurodegenerative disease is: what accounts for the extreme tissue and cell-type specificity of disease pathology? The offending protein is often expressed ubiquitously, yet parts of the brain are disproportionately affected. Moreover, some neurodegenerative conditions, like Alzheimer's, can come and go with the patient experiencing "good days" when their cognitive functions are nearly intact and "bad days" when the effects of the disease are apparent. This suggests that the neural functionality is partially intact, but that the neural network is unable to make proper use of it.
In order to begin to explore this question, we are beginning to transfer our cell biological understanding of things that happen during aggregation-induced toxicity into an animal model where we can look at aggregation in functional neurons. |
|
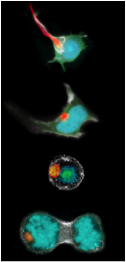

Harnessing the Dark Side of Protein FoldingBecause of its strong association with disease pathology, we have come to regard aggregation as a deleterious process that is ideally to be avoided. Our recent research has led us to see aggregation in a new light: not as a toxic process one ought to avoid, but as another side of protein folding that we have to harness. In exploring the mechanism behind what we call “aggregation quality control” we have realized that aggregation is a normal, homeostatic, and often protective process that the cell engages in during non-stressed times and during times of stress in order to more effectively manage the complicated biology of protein folding. Our goal in continuing to dissect this pathway is to exert control over aggregation quality control as a strategy for more effective protein production and a potential therapeutic strategy in dealing with toxic aggregation.
Spatial organization of protein folding quality control in the cytosol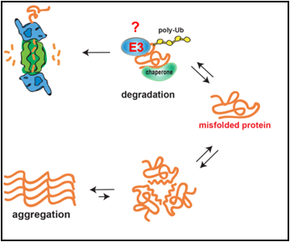
Cellular viability depends on timely and efficient management of proteins that misfold due to damage, stress, mutations, and aging. We use the power of yeast genetics to discover novel players in several stages of this pathway. We want to know: How is a misfolded protein recognized? How is a misfolded protein ubiquitinated? How is the triage decision made to refold, degrade, or aggregate a misfolded protein? How are aggregates delivered to inclusions? JUNQ and IPOD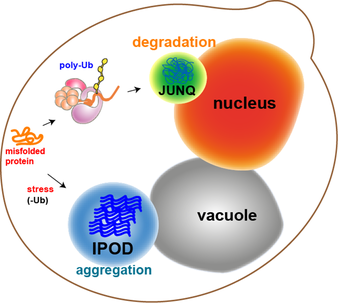 Yeast
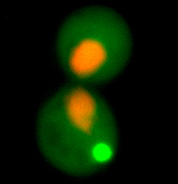
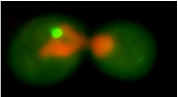
We recently discovered that there are at least two different types of inclusions in eukaryotic cells: JUNQs and IPODs.
JUNQ and IPOD have different functions. The JUNQ (juxta-nuclear quality control compartment) is the cellular quality control center where soluble aggregates from the cytosol and the ER accumulate for proteasomal degradation and re-folding. The IPOD (insoluble protein deposit) sequesters harmful aggregates and amyloids from the rest of the cytosol, thereby serving a protective function. Cells partition their misfolded protein load into one of these two compartments, which also exist under normal conditions. In order to go to the JUNQ a protein must be a ubiquitinated quality control substrate. Proteins that can't be ubiquitinated are instead sent to the IPOD, where they undergo active aggregation. |
|

The protein folding problem
The common denominator of all proteins in the cell is their need to maintain a set of low-energy conformations of their folding structure, known as the native state. Folding to the native state is essential to protein function, and proteins that fail to do so pose a threat to the cellular environment by exhibiting a tendency to misfold, aggregate, and otherwise disrupt the cellular environment. Roughly speaking, folding to the native state involves the burial of hydrophobic amino acids inside the core of the protein, and the exposure of hydrophilic amino acids on the outside of the protein. Our understanding of the relationship between amino acid sequence and protein structure are complicated, however, by several factors:
1. proteins in the cell are not in an ideal aqueous environment - crowding effects and local nano-environments will tend to dominate thermodynamic considerations involved in folding. 2. protein function and protein-protein interaction is driven by hydrophobic residues that are not buried in the core of the protein. 3. protein structure is dynamic - even in the native state a protein may sample multiple conformations. What interests us is the question: what is the difference between a native protein and a misfolded protein from the standpoint of the cell and its protein folding quality control machinery? We approach this problem in collaboration with the lab of Jeremy England, who has recently developed a ground-breaking theoretical bio-physical model for protein misfolding. With out cell biological tools and in vivo models of protein misfolding, we can now apply Jeremy's theoretical approaches to protein misfolding problems in the cytosol of a living cell. Our joint aim is to understand the following questions: How does the cell recognize a protein as being misfolded (as opposed to a natively unstructured protein, a protein that has a dynamic structure with multiple allosteric conformations, or a completely unfolded protein)? How does the cell distinguish between misfolded proteins that should be degraded or refolded and amyloidogenic proteins that have no hope of folding and should be taken to an aggregate inclusion? |
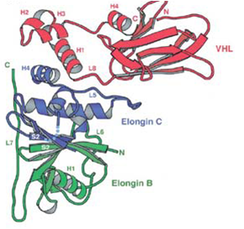
The von-Hippel Lindau protein (VHL) is a tumor suppressor that has been extensively studied as a model for protein misfolding. The VHL protein (red) is constitutively misfolded unless it can bind to its binding partners Elongin B/C (blue and green). Several tumoragenic mutations disrupt the binding and lead to VHL misfolding and degradation by the protein folding quality control machinery of the cell.
In collaboration with the lab of Jeremy England, we are studying the relationship between VHL sequence and its tendency to misfold. Our first results from this project were published in Brock et al., Structure 2015. |
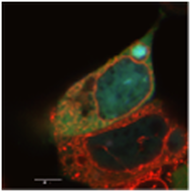
What is the function of aggregate inclusions in the cell?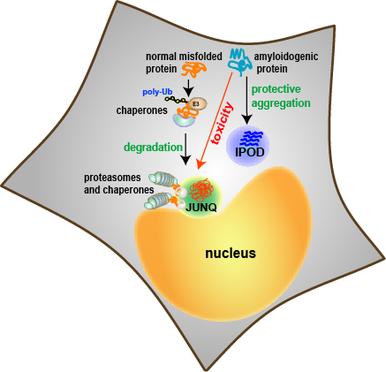 Mammalian cells
Aggregate inclusions are the common cell biological feature of many neurodegenerative diseases. These diseases are caused by proteins that don't share sequence homology, structural similarities, or functional features, but the thing they have in common is a tendency to aggregate and form inclusions.
Far from being random deposits of insoluble materials, inclusions are now understood to be an integral part of the cellular response to protein aggregation. Hence, aggregated proteins are delivered to inclusions in an active and directed manner, requiring energy and cellular machines. |
|
The role of aging in protein aggregation and neurodegenerative disease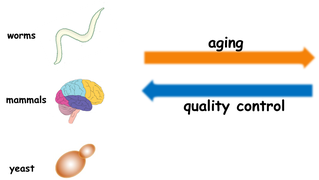
In a variety of organisms, from bacteria to mammals, aging is regulated by a genetic pathway. Protein folding quality control seems to decline with aging. Conversely, delaying aging improves the ability of the organism to deal with protein misfolding and protein damage. This is especially relevant given that so many diseases caused by problems with quality control and aggregation are triggered by aging.
|
|

